タクザイロ皮下注300mgシリンジ 発売中
海外第Ⅲ相臨床試験 長期投与試験 【海外データ】
「禁忌を含む注意事項等情報」は電子添文をご参照ください。
海外第Ⅲ相長期投与試験(DX-2930-04試験)1,2)
1)社内資料(承認時評価資料):海外第Ⅲ相多施設共同長期投与試験
2)Banerji A, et al. Allergy. 2022; 77(3): 979-990.(supplement)
本試験はShire社(現:Takeda)の資金提供により実施された。
本論文の著者のうち3名はTakedaの社員である。
著者に同社よりコンサルタント料等を受領している者が含まれる。
試験概要
目的
Ⅰ型又はⅡ型の遺伝性血管性浮腫(HAE)患者を対象として、急性発作予防におけるタクザイロの長期安全性及び有効性を検討する。
対象
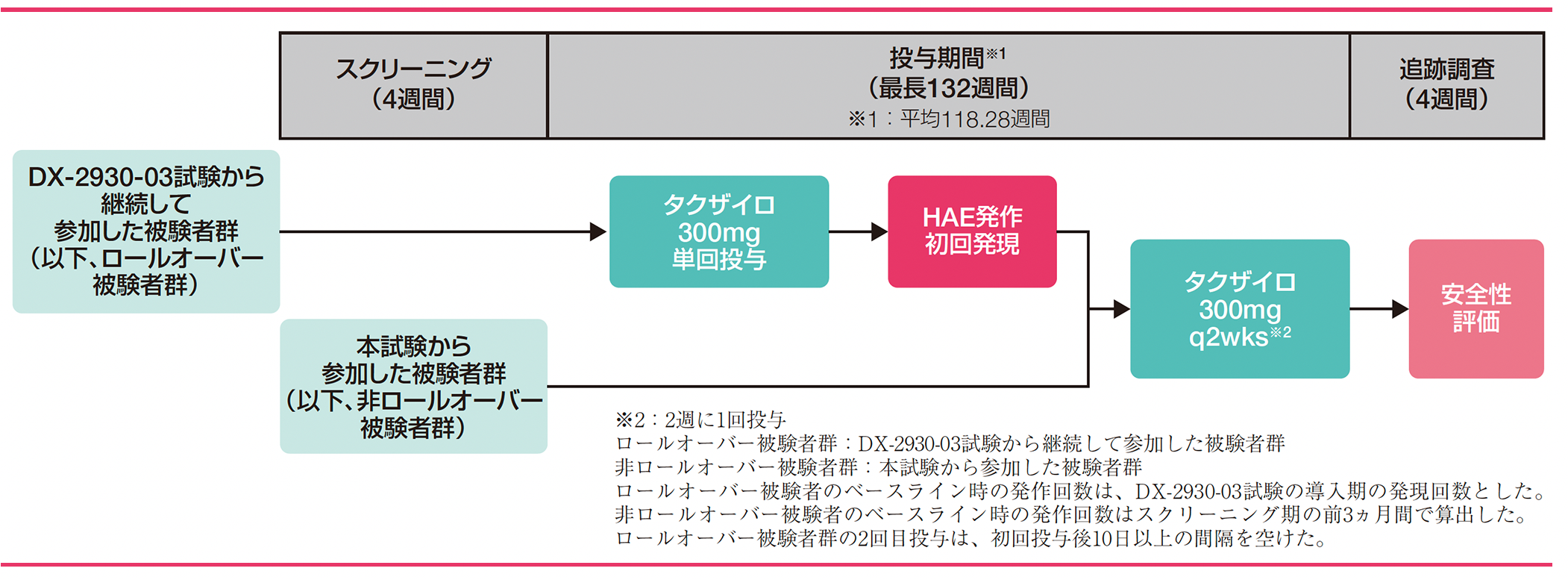
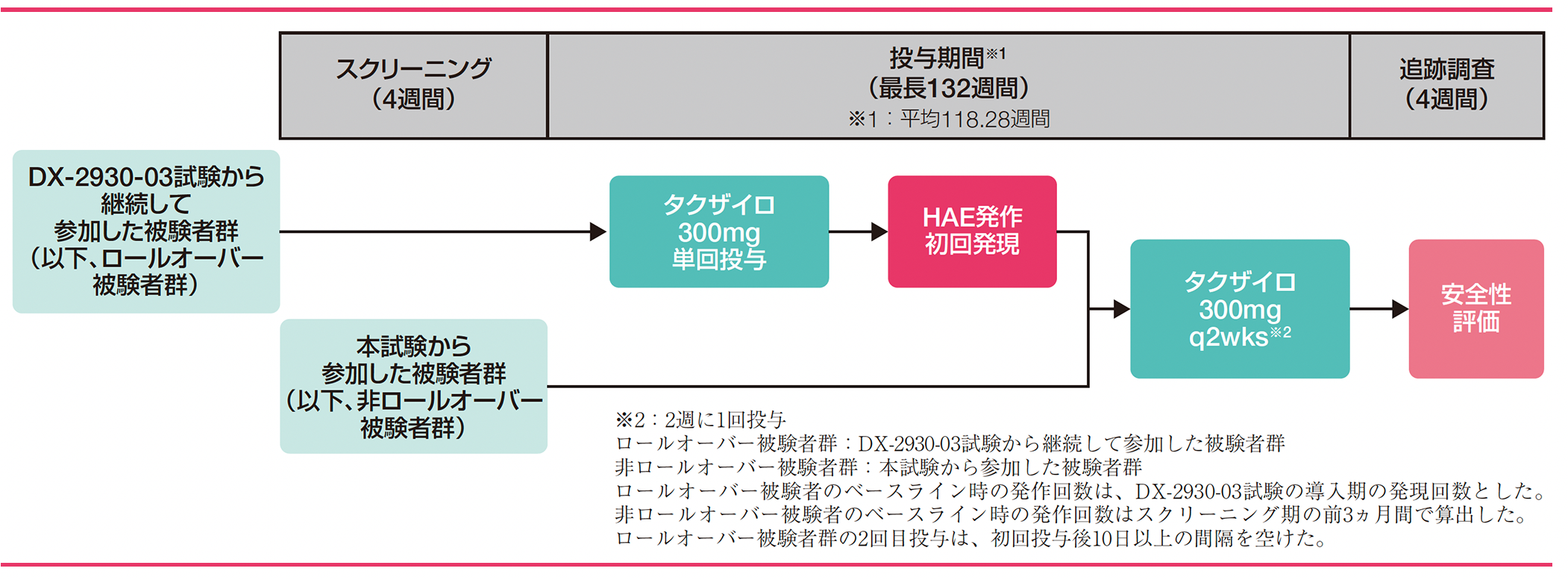
先行した多施設共同第Ⅲ相試験(DX-2930-03試験)のDay182に二重盲検下の投与期間を完了し、長期投与試験への参加に同意した被験者群(ロールオーバー被験者群)109例及びDX-2930-03試験に参加していない被験者(非ロールオーバー被験者群)103例
評価項目
安全性評価項目:有害事象、治療継続率 等
副次評価項目:投与期間中に治験責任医師が確認したHAE発作発現回数とベースラインからの平均低下率、HAE発作発現回数のベースラインからの平均低下率推移(1~33ヵ月評価時点まで) 等
探索的評価項目:投与期間中における無発作日数及び無発作日数の割合・無発作期間の平均日数・最大無発作期間 等
その他の評価項目:血管性浮腫に伴うQoL (AE-QoL)質問票各スコアの変化量 等
試験デザイン
患者背景
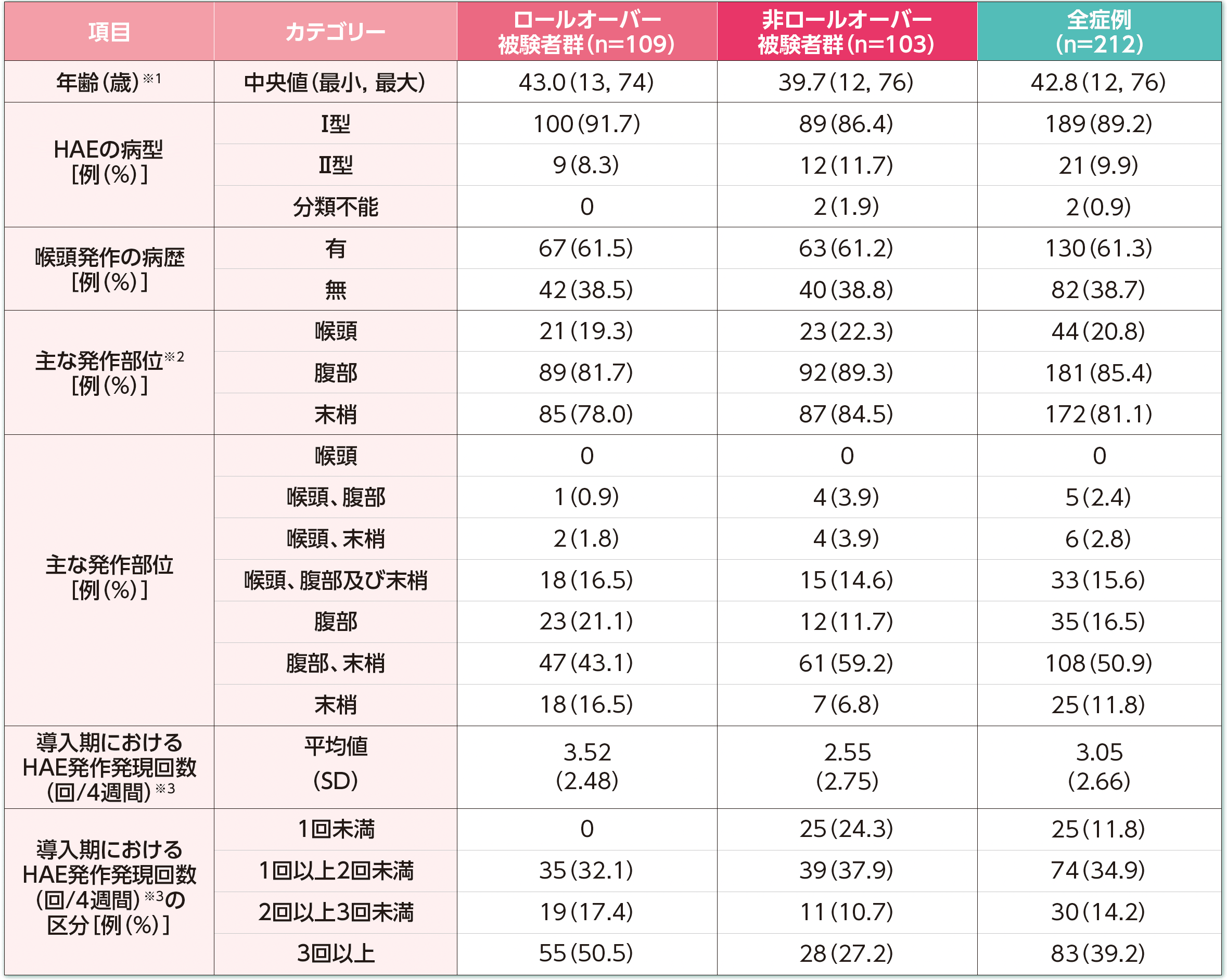
※1:年齢は、生年月日とインフォームドコンセントの日付との差として算出し、年数で切り捨てた。
※2:複数のカテゴリーに重複してカウントした症例を含む。
※3:ロールオーバー被験者群はDX-2930-03試験の導入期で治験責任医師が確認したHAE発作発現回数、非ロールオーバー被験者群は病歴で報告された期間におけるHAE発作発現回数を、導入期の日数又は病歴で報告された期間の日数でそれぞれ除し、28日を乗じて算出した。非ロールオーバー被験者群では、スクリーニング前3ヵ月間の病歴上の発作発現回数を使用した。
HAE:遺伝性血管性浮腫、SD:標準偏差
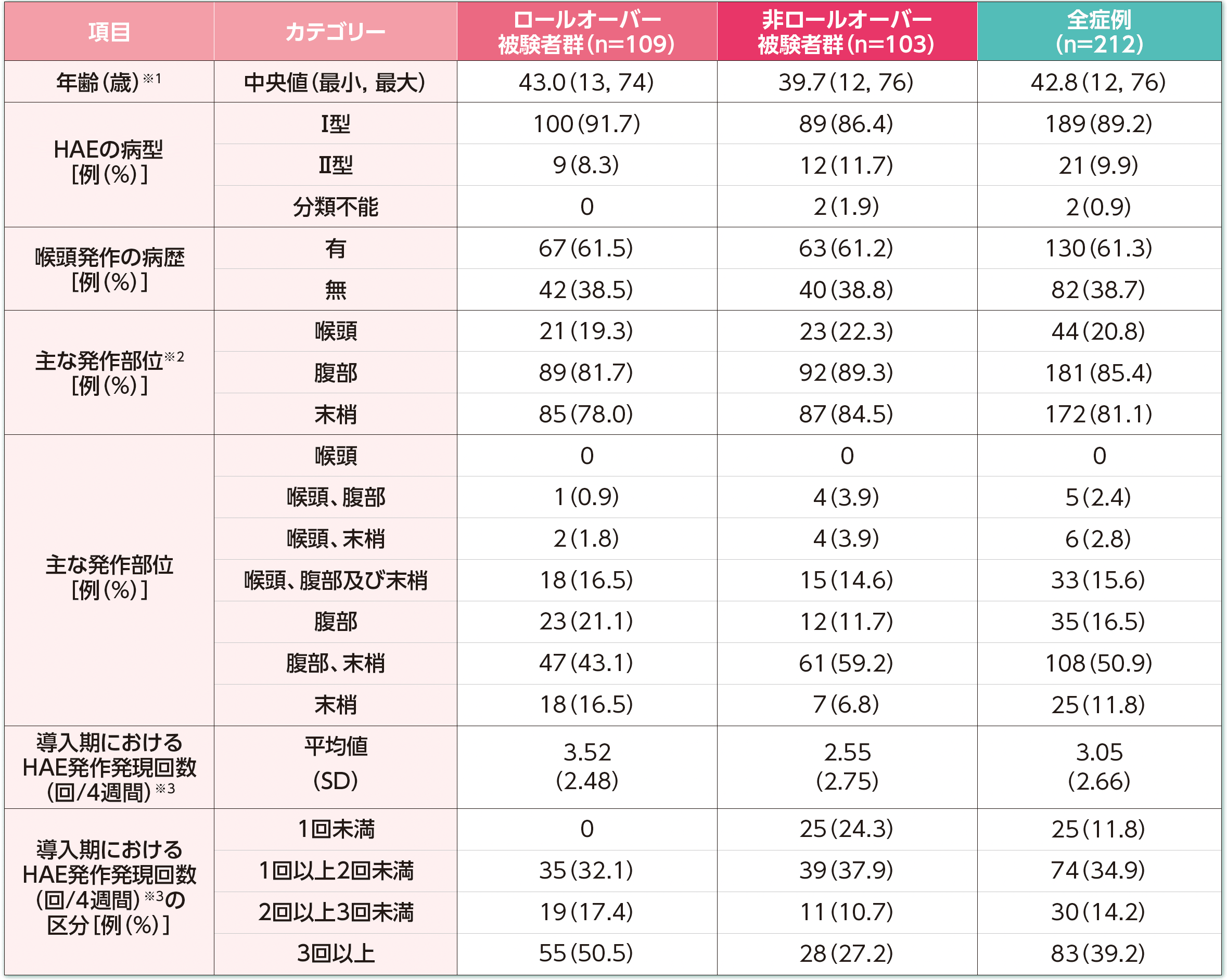
※1:年齢は、生年月日とインフォームドコンセントの日付との差として算出し、年数で切り捨てた。
※2:複数のカテゴリーに重複してカウントした症例を含む。
※3:ロールオーバー被験者群はDX-2930-03試験の導入期で治験責任医師が確認したHAE発作発現回数、非ロールオーバー被験者群は病歴で報告された期間におけるHAE発作発現回数を、導入期の日数又は病歴で報告された期間の日数でそれぞれ除し、28日を乗じて算出した。非ロールオーバー被験者群では、スクリーニング前3ヵ月間の病歴上の発作発現回数を使用した。
HAE:遺伝性血管性浮腫、SD:標準偏差
安全性
1. 有害事象の発現頻度(HAE発作関連事象を除く)(安全性評価項目)
治験薬との因果関係が否定できない有害事象の発現頻度は、ロールオーバー被験者群で45.9%(50/109例)、非ロールオーバー被験者群で64.1%(66/103例)でした。
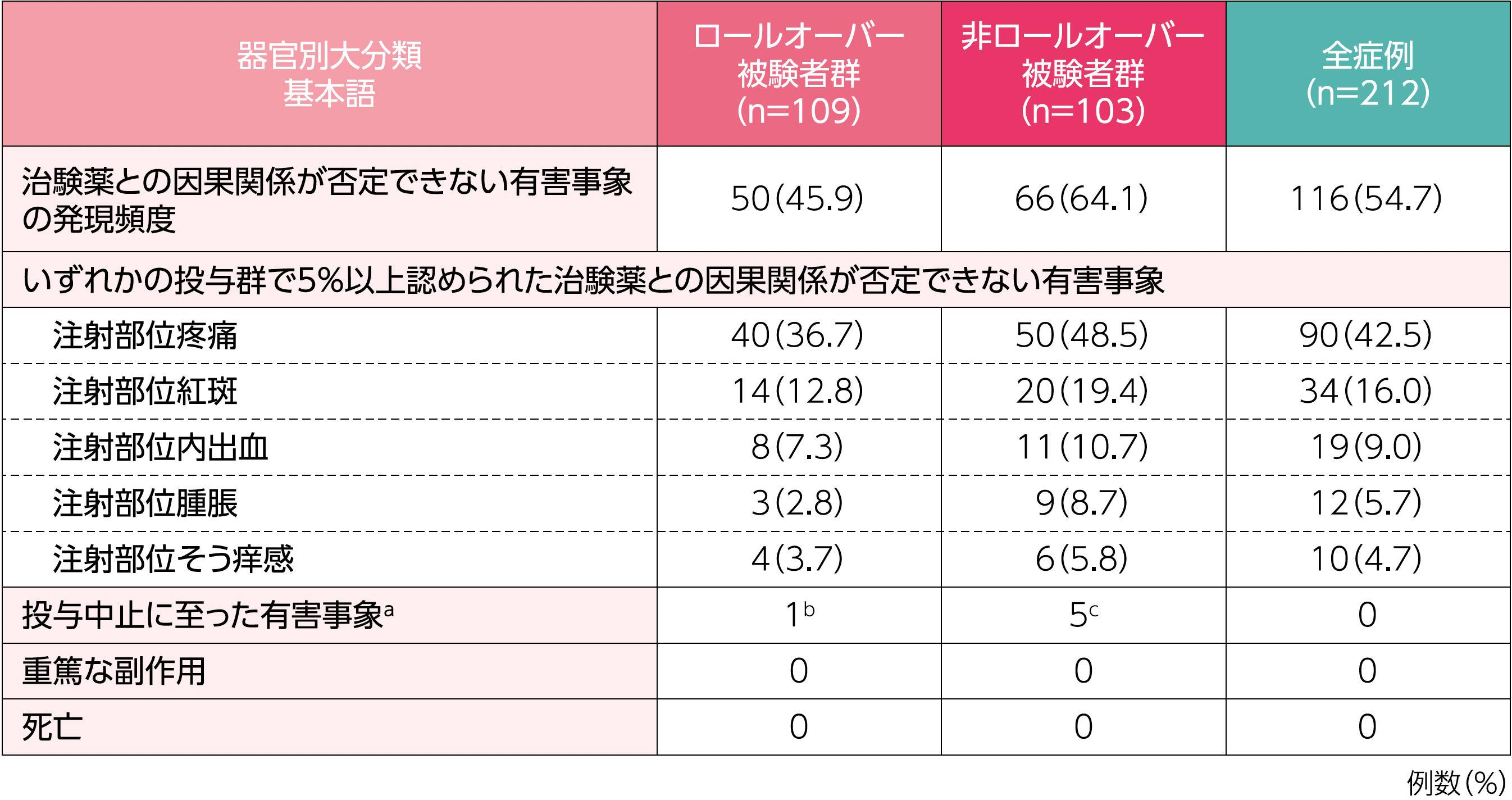
a:転帰は肺炎(不明)を除いていずれも回復であり、治験薬と因果関係ありと判定されたのは過敏症3例であった(いずれも非重篤)。
b:上部消化管出血及び肺炎
c:過敏症3例、ALT増加及びAST増加1例、ALT増加、AST増加及び血中クレアチンホスホキナーゼ増加1例
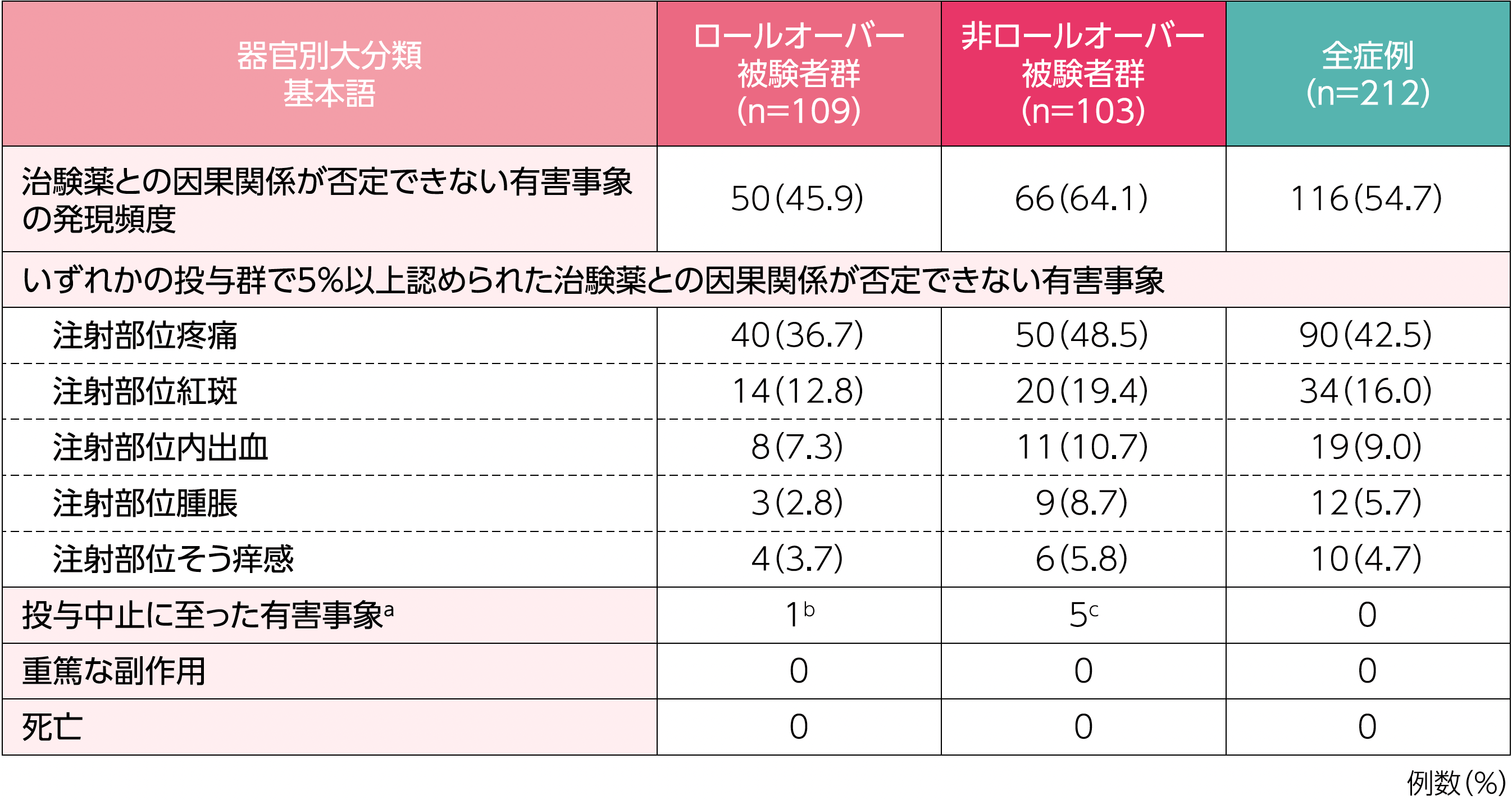
a:転帰は肺炎(不明)を除いていずれも回復であり、治験薬と因果関係ありと判定されたのは過敏症3例であった(いずれも非重篤)。
b:上部消化管出血及び肺炎
c:過敏症3例、ALT増加及びAST増加1例、ALT増加、AST増加及び血中クレアチンホスホキナーゼ増加1例
2. 治療継続率(安全性評価項目)
本試験において投与期間を完了した被験者の割合は26.4%(56/212例)であり、その内訳は、ロールオーバー被験者群28.4%(31/109例)、非ロールオーバー被験者群24.3%(25/103例)でした。試験を中止した156例のうち117例は本剤が市販で利用可能となり被験者自身の意思で市販製剤に移行し、被験者の81.6%(173/212例)は試験を完了又は市販製剤へ移行しました。また、173例(81.6%)は少なくとも30ヵ月の投与を完了していました。
有効性
1. 投与期間中に治験責任医師が確認したHAE発作発現回数とベースラインからの平均低下率(副次評価項目)
ロールオーバー被験者群、非ロールオーバー被験者群及び全症例におけるHAE発作発現回数の平均値(SD)は、ベースラインではそれぞれ4週間あたり3.52回(2.48)、2.55回(2.75)及び3.05回(2.66)であり、投与期間ではそれぞれ4週間あたり0.27回(0.58)、0.22回(0.52)及び0.25回(0.55)でした。ベースラインからのHAE発作低下率(SD)はそれぞれー92.38%(12.81)、ー81.96%(96.76)及びー87.37%(67.72)でした。
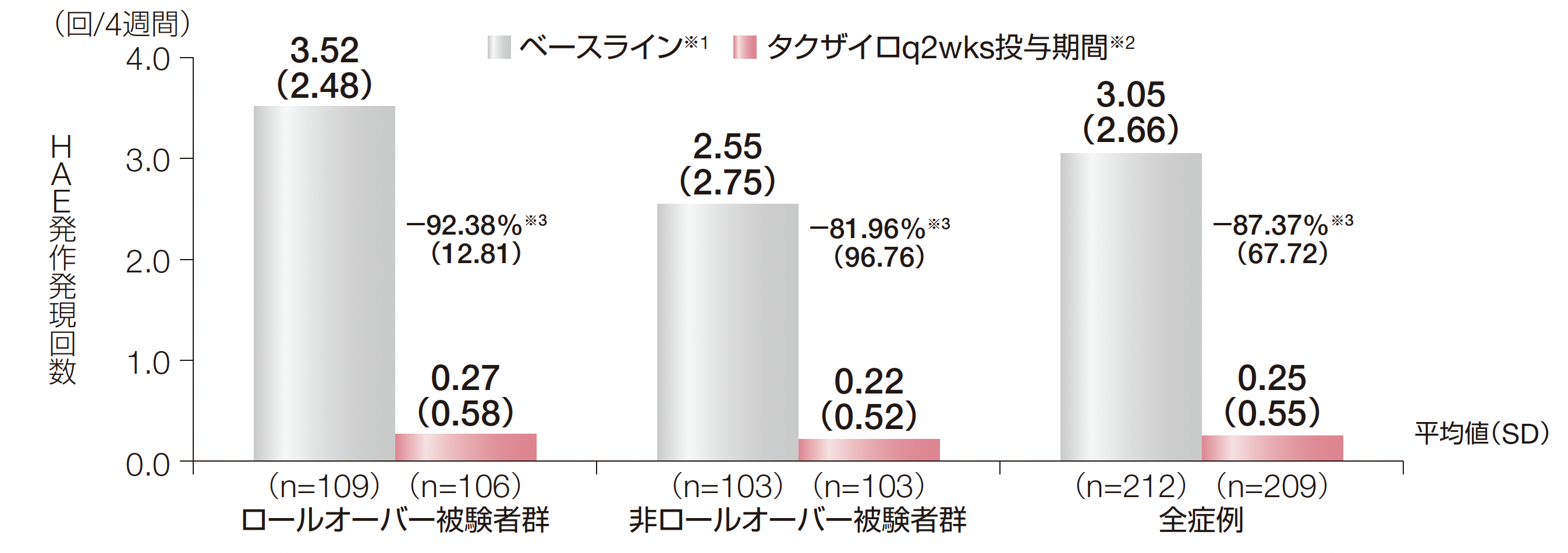
※1:ロールオーバー安全性解析対象集団におけるベースラインのHAE発作発現回数は、DX-2930-03試験の導入期間に治験責任医師が確認した
HAE発作発現回数を導入期間の日数で除し、28日を乗じた。非ロールオーバー安全性解析対象集団におけるベースラインのHAE発作発現回数は、スクリーニング前3ヵ月間のHAE発作発現回数をその観察日数で除し、28日を乗じた。
※2:投与期間中に治験責任医師が確認したHAE発作発現回数は、被験者ごとに、投与期間(ロールオーバー被験者群では定期投与期間)に治験責任医師が確認したHAE発作発現回数をその投与期間の日数で除し、28日を乗じた。
※3:4週間あたりのHAE発作発現回数のベースラインからの低下率は、100%×(4週間あたりのHAE発作発現回数-ベースラインのHAE発作発現回数)/ベースラインのHAE発作発現回数で算出した(症例数:ロールオーバー被験者群106例、非ロールオーバー被験者群98例)。
ロールオーバー被験者群の定期投与期間は、2回目の治験薬投与時からDay 924の来院又は早期中止日の早い日までとした。
HAE:遺伝性血管性浮腫、SD:標準偏差
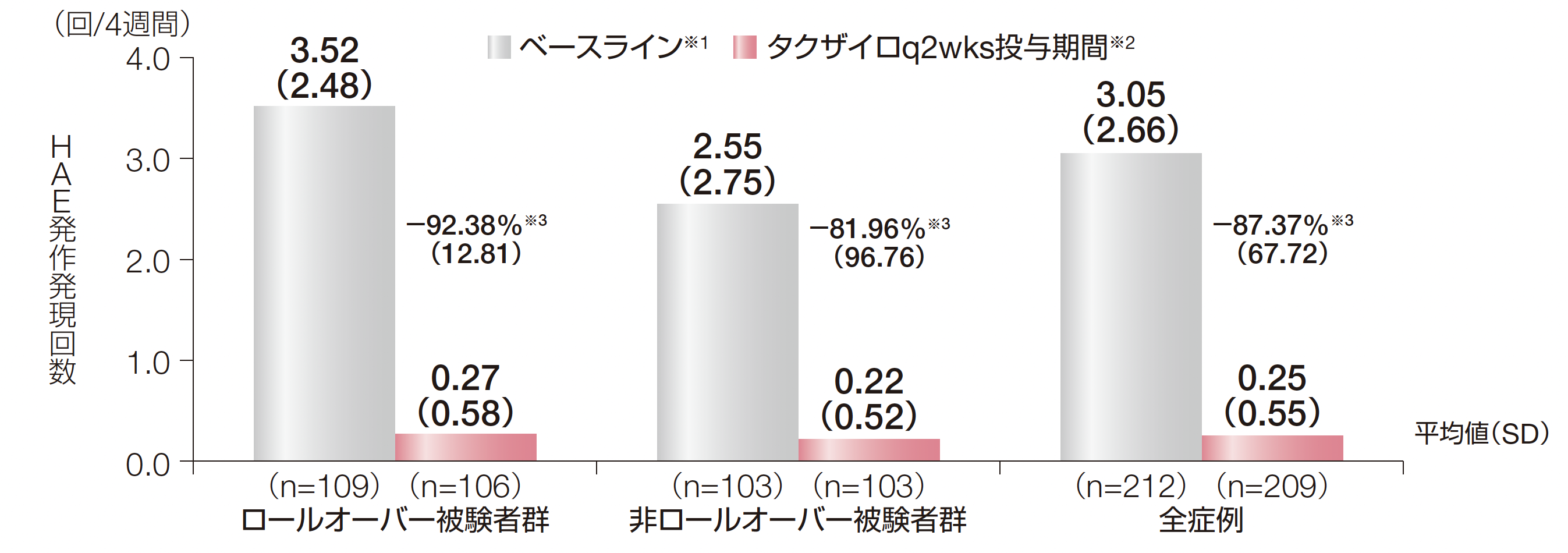
※1:ロールオーバー安全性解析対象集団におけるベースラインのHAE発作発現回数は、DX-2930-03試験の導入期間に治験責任医師が確認した
HAE発作発現回数を導入期間の日数で除し、28日を乗じた。非ロールオーバー安全性解析対象集団におけるベースラインのHAE発作発現回数は、スクリーニング前3ヵ月間のHAE発作発現回数をその観察日数で除し、28日を乗じた。
※2:投与期間中に治験責任医師が確認したHAE発作発現回数は、被験者ごとに、投与期間(ロールオーバー被験者群では定期投与期間)に治験責任医師が確認したHAE発作発現回数をその投与期間の日数で除し、28日を乗じた。
※3:4週間あたりのHAE発作発現回数のベースラインからの低下率は、100%×(4週間あたりのHAE発作発現回数-ベースラインのHAE発作発現回数)/ベースラインのHAE発作発現回数で算出した(症例数:ロールオーバー被験者群106例、非ロールオーバー被験者群98例)。
ロールオーバー被験者群の定期投与期間は、2回目の治験薬投与時からDay 924の来院又は早期中止日の早い日までとした。
HAE:遺伝性血管性浮腫、SD:標準偏差
2. HAE発作発現回数のベースラインからの平均低下率推移
(1~33ヵ月評価時点まで)(副次評価項目)
ロールオーバー被験者群、非ロールオーバー被験者群における投与期間1~33ヵ月までのHAE発作発現回数のベースラインからの平均低下率(SD)は、それぞれ1ヵ月時点で-88.72%(25.16)、-77.84%(72.62)、2ヵ月時点で-91.42%(25.38)、-81.12%(98.31)、33ヵ月時点で-98.60%(7.00)、-96.85%(14.36)でした。
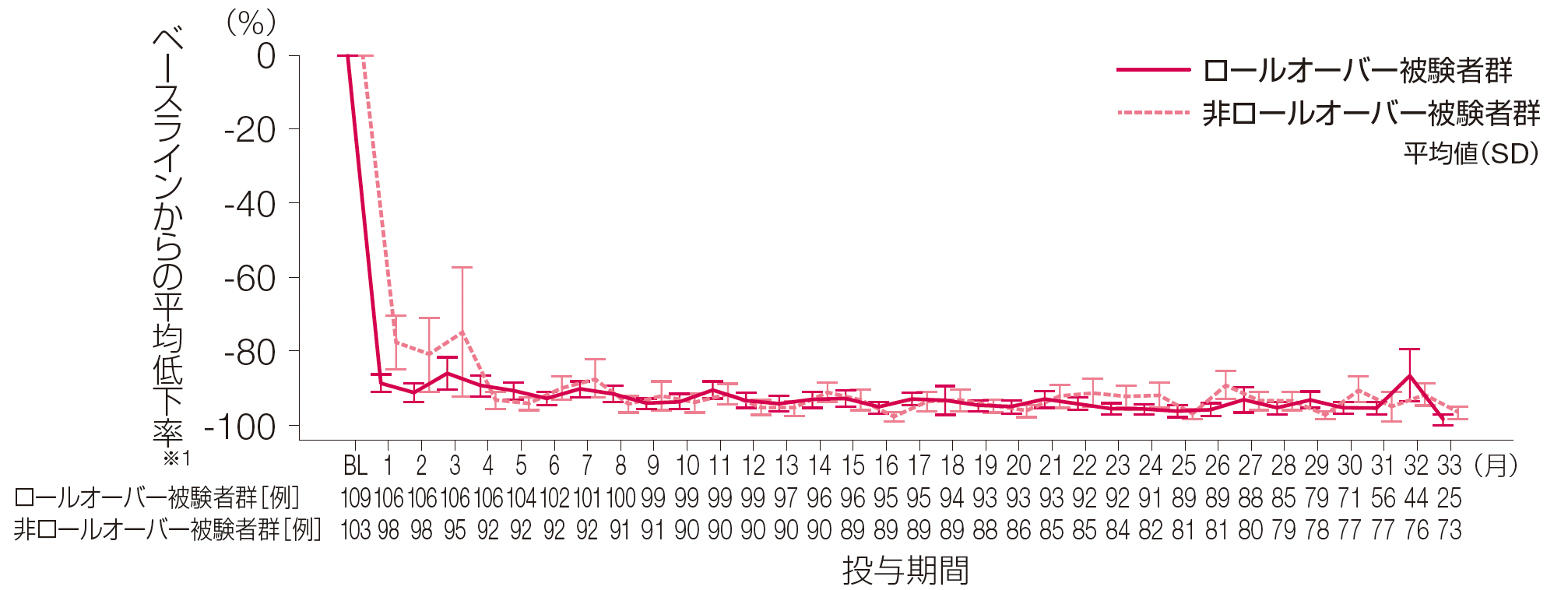
※1: 4週間あたりのHAE発作発現回数のベースラインからの低下率は、100%×(4週間あたりのHAE発作発現回数-ベースラインのHAE発作発現回数)/ベースラインのHAE発作発現回数で算出した。
BL:ベースライン、HAE:遺伝性血管性浮腫、SD:標準偏差
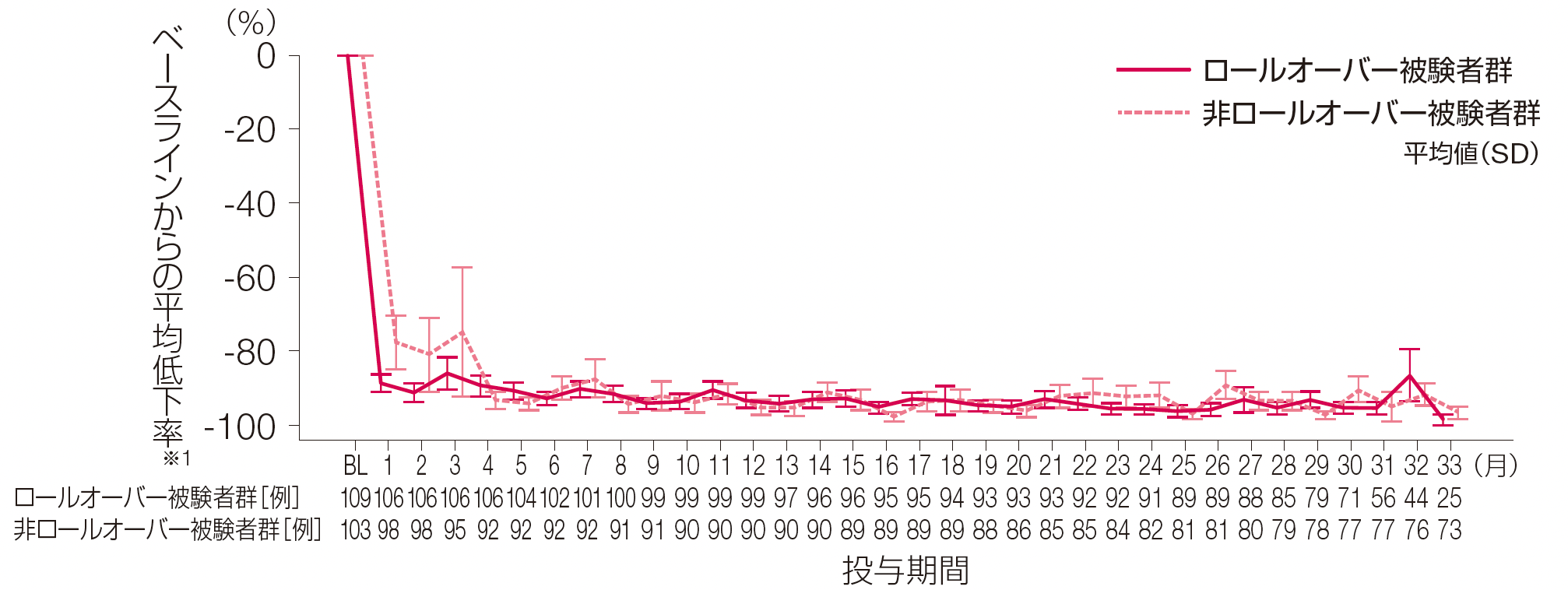
※1: 4週間あたりのHAE発作発現回数のベースラインからの低下率は、100%×(4週間あたりのHAE発作発現回数-ベースラインのHAE発作発現回数)/ベースラインのHAE発作発現回数で算出した。
BL:ベースライン、HAE:遺伝性血管性浮腫、SD:標準偏差
3. 投与期間における無発作日数(探索的評価項目)
ロールオーバー被験者群における無発作期間の平均値(SD)は380.7日(319.3)、非ロールオーバー被験者群における無発作期間の平均値(SD)は450.4日(369.9)でした。
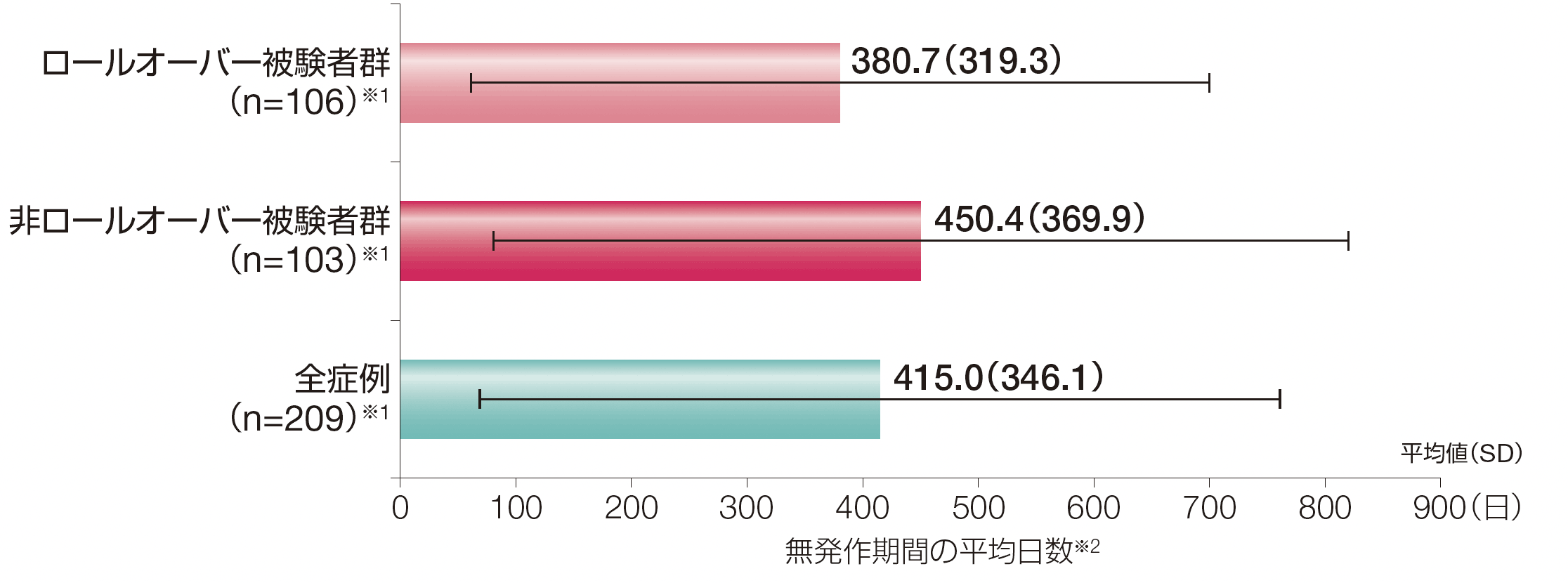
※1:導入期間中に発作を1回以上発現した被験者数
※2:無発作期間の平均日数は、被験者の無発作期間の平均値から算出した。
被験者の合計定期投与期間は、ロールオーバー被験者群で2935.9ヵ月、非ロールオーバー被験者群で3005.4ヵ月、全症例で5941.3ヵ月であった。
SD 標準偏差
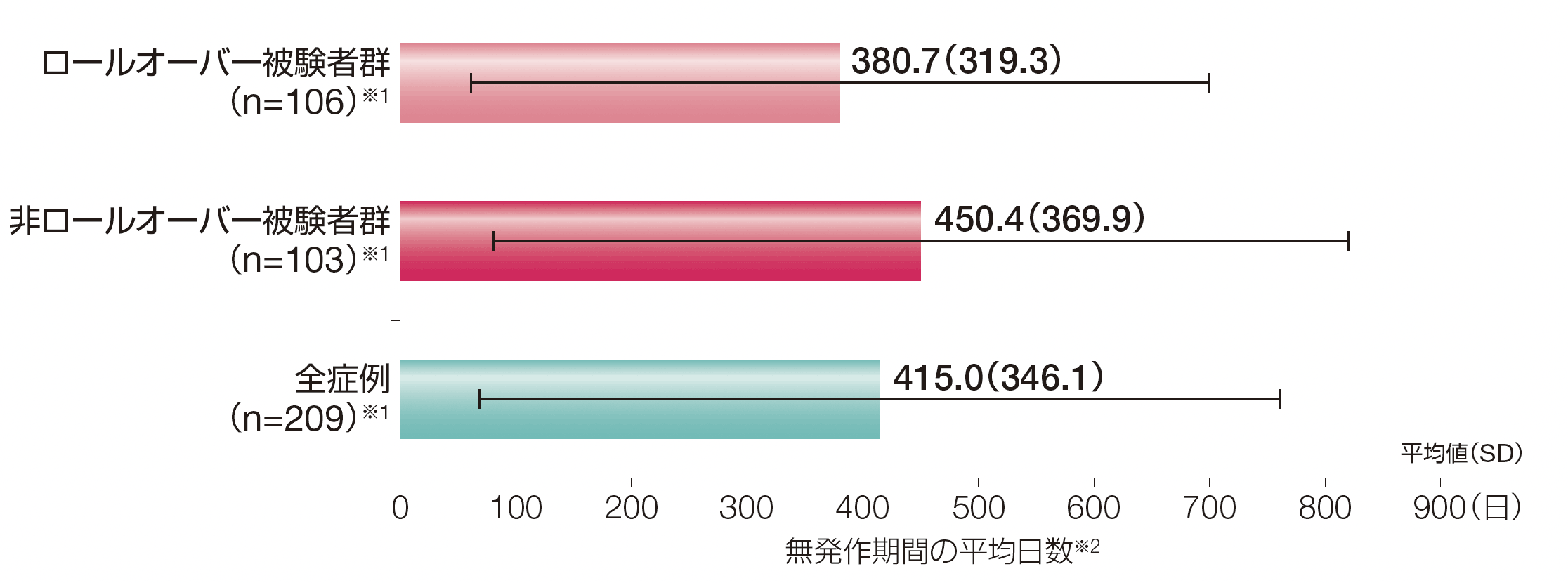
※1:導入期間中に発作を1回以上発現した被験者数
※2:無発作期間の平均日数は、被験者の無発作期間の平均値から算出した。
被験者の合計定期投与期間は、ロールオーバー被験者群で2935.9ヵ月、非ロールオーバー被験者群で3005.4ヵ月、全症例で5941.3ヵ月であった。
SD 標準偏差
参考情報
AE-QoL質問票各スコアの変化量(その他の評価項目)
ロールオーバー被験者群及び非ロールオーバー被験者群のAE-QoL質問票の総スコア及び4つの領域スコア[機能、疲労/気分、恐怖/羞恥及び栄養(食事)]のDay 0〜最終来院までの変化量の平均値(SD)は以下のとおりでした。
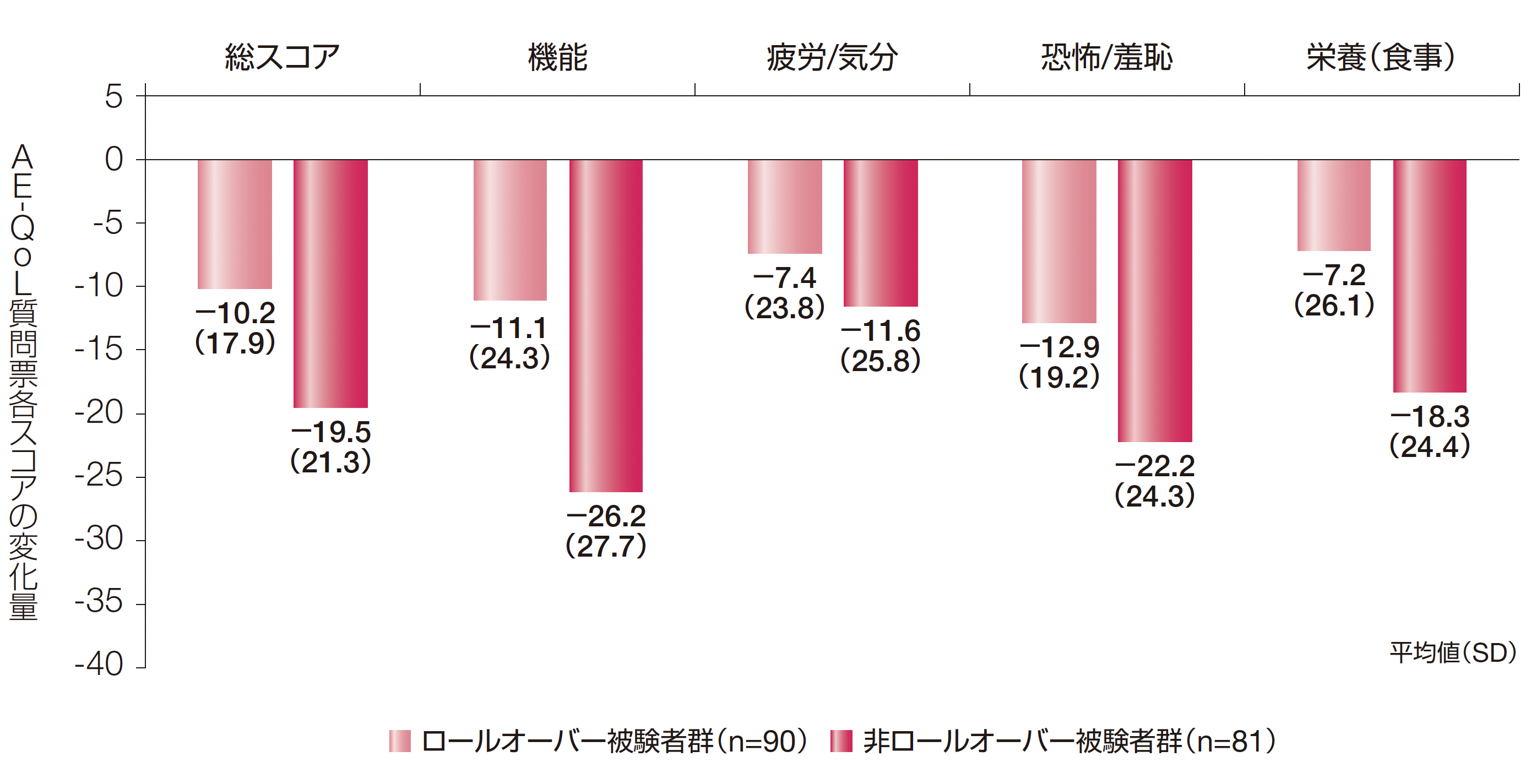
AE-QoL質問票は血管性浮腫に特化した自己記入式のQoL評価ツールであり、質問票の17項目を1(まったくなし)~ 5(非常に多い)の5段階で評価する。各項目のスコアを集計し、総スコア及び4つの領域スコア[機能、疲労/気分、恐怖/羞恥及び栄養(食事)]を求めた。得られた領域スコア(当該領域に含まれる項目スコアの平均値)及び総スコア(全ての項目スコアの平均値)を、一次変換で最終的なパーセンテージスコア(0 ~100)に変換した。スコアが大きいほどQoLが低いことを示す。
AE-QoL:血管性浮腫に伴うQoL、SD:標準偏差
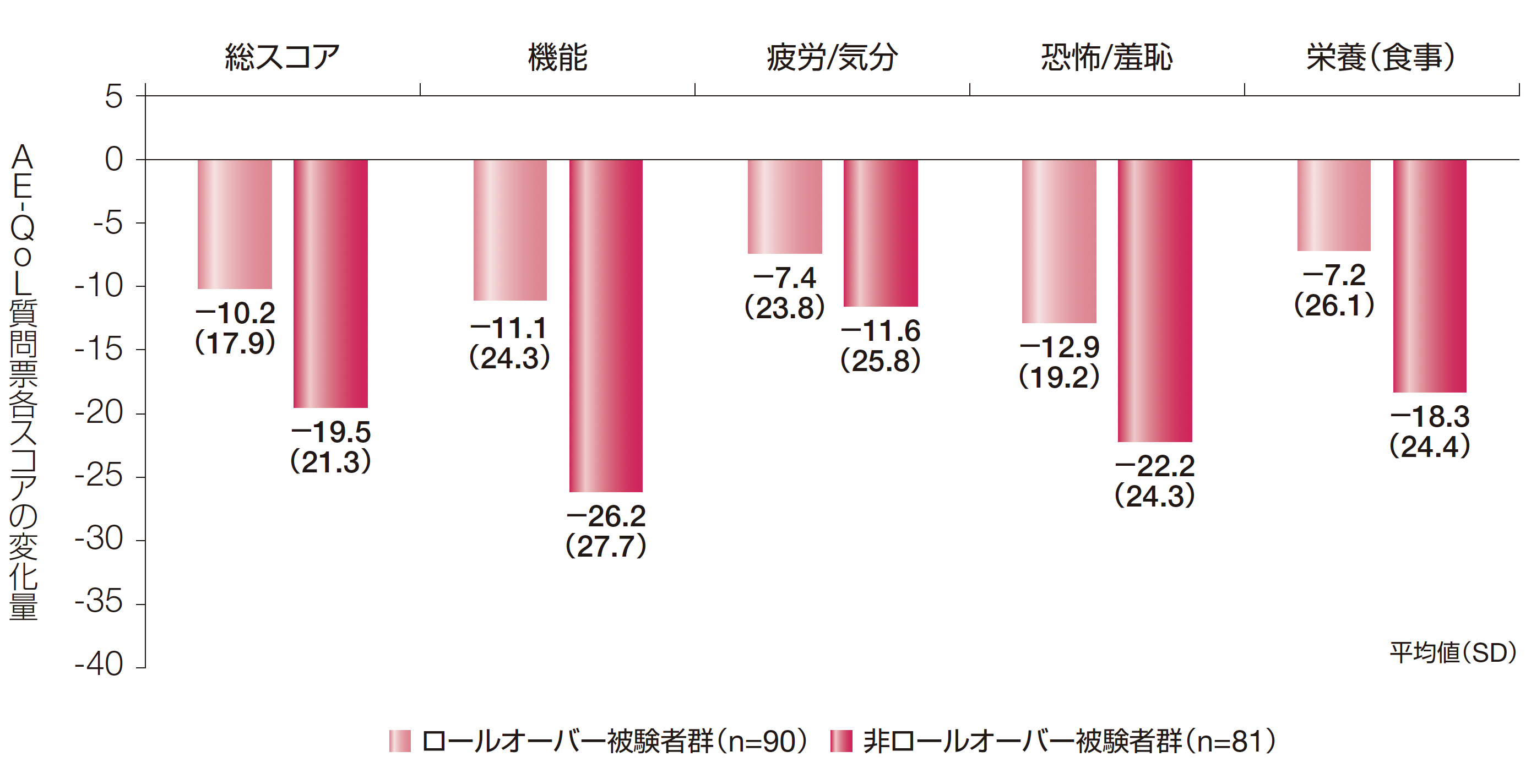
AE-QoL質問票は血管性浮腫に特化した自己記入式のQoL評価ツールであり、質問票の17項目を1(まったくなし)~ 5(非常に多い)の5段階で評価する。各項目のスコアを集計し、総スコア及び4つの領域スコア[機能、疲労/気分、恐怖/羞恥及び栄養(食事)]を求めた。得られた領域スコア(当該領域に含まれる項目スコアの平均値)及び総スコア(全ての項目スコアの平均値)を、一次変換で最終的なパーセンテージスコア(0 ~100)に変換した。スコアが大きいほどQoLが低いことを示す。
AE-QoL:血管性浮腫に伴うQoL、SD:標準偏差
試験概要(詳細)
目的
Ⅰ型又はⅡ型の遺伝性血管性浮腫(HAE)患者を対象として、急性発作予防におけるタクザイロの長期安全性及び有効性を検討する。
対象
先行した多施設共同第Ⅲ相試験(DX-2930-03試験)のDay 182に二重盲検下の投与期間を完了し、長期投与試験への参加に同意した被験者群(ロールオーバー被験者群)109例及びDX-2930-03試験に参加していない被験者群(非ロールオーバー被験者群)103例
試験方法
12歳以上でHAE(Ⅰ型又はⅡ型)の確定診断を受け、導入期間中にHAE発作を1回以上発現した被験者209例を対象に、タクザイロ300mgを2週に1回反復投与した。最長132週間の投与期間終了後、4週間の追跡調査を実施し、全被験者の安全性評価を実施した。
評価項目
安全性評価項目:有害事象、治療継続率 等
副次評価項目:投与期間中に治験責任医師が確認したHAE発作発現回数とベースラインからの平均低下率、HAE発作発現回数のベースラインからの平均低下率推移(1〜33ヵ月評価時点まで) 等
探索的評価項目:投与期間中における無発作日数及び無発作日数の割合・無発作期間の平均日数・最大無発作期間 等
その他の評価項目:血管性浮腫に伴うQoL (AE-QoL)質問票各スコアの変化量 等
解析計画
有効性解析は、安全性解析対象集団(治験薬の投与を1回以上受けた全ての被験者)を用いて実施した。4週間あたりのHAE発作発現回数は各解析対象集団別に4週間ごとに記述的に要約し(1ヵ月は28日間と定義)、非ロールオーバー被験者群では投与期間全体で示し、ロールオーバー被験者群では投与期間のうち定期投与期間(2回目の投与以降の期間)で示した。HAE無発作日数(HAE発作が発現しなかった日と定義)の割合は、無発作日を投与期間の日数で除し、算出した。無発作期間(治験責任医師が確認した無発作の連続日数と定義)は、被験者ごとの無発作期間の平均値から算出した。有害事象は、安全性解析対象集団、ロールオーバー安全性解析対象集団及び非ロールオーバー安全性解析対象集団を用いて要約した。その他の解析として、AE-QoL質問票の各項目に対する回答及びAE-QoL総スコア及び4つの領域スコア[機能、疲労/気分、恐怖/羞恥及び栄養(食事)]について要約した。